Saúde
Estudo abre novas possibilidades de tratamento para forma de autismo
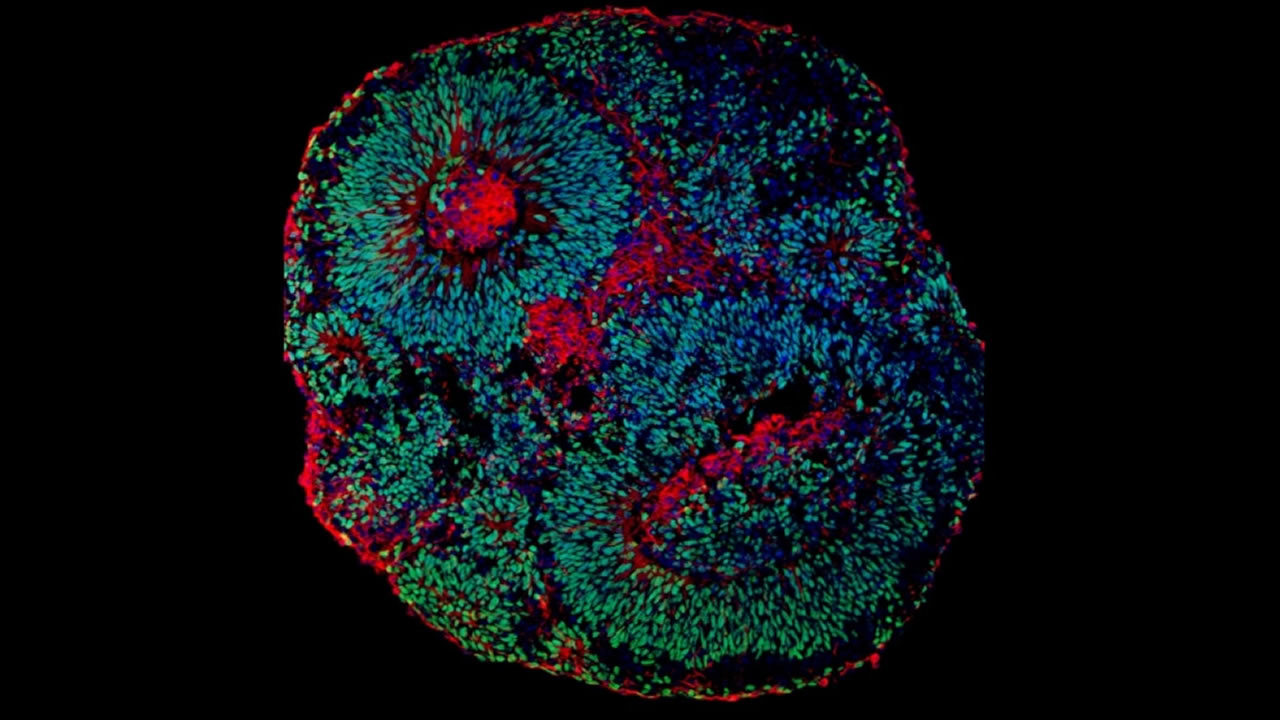
André Julião | Agência FAPESP – Um grupo liderado por cientistas brasileiros da Universidade Estadual de Campinas (Unicamp) e da Universidade da Califórnia San Diego, nos Estados Unidos, desvendou o mecanismo causador da síndrome de Pitt-Hopkins, disfunção neuropsiquiátrica que tem características de transtorno do espectro autista (TEA). Além disso, os pesquisadores conseguiram reverter a evolução da síndrome em modelos de laboratório, abrindo novas possibilidades de tratamento.
O trabalho, apoiado pela FAPESP, foi publicado nesta segunda-feira (02/05) na revista Nature Communications.
“Para a maioria dos casos de TEA, não se sabe qual gene causa a condição quando mutado. Assim é também para a maioria das doenças neuropsiquiátricas, como esquizofrenia, depressão e transtorno bipolar. A síndrome de Pitt-Hopkins, por sua vez, tem como origem uma mutação no gene TCF4. Mas, até então, não eram conhecidos seus mecanismos moleculares, ou seja, o que há de diferente nas células do sistema nervoso dos pacientes com a mutação”, conta Fabio Papes, professor do Instituto de Biologia (IB-Unicamp) e um dos coordenadores do estudo.
O grupo liderado por Papes e pelo professor da Universidade da Califórnia San Diego Alysson Muotri, no entanto, foi além da descoberta do mecanismo causador da condição.
Os cientistas testaram maneiras de interferir na evolução do quadro e conseguiram reverter os efeitos causados pela mutação. O sucesso obtido nos experimentos abre caminho para o desenvolvimento tanto de medicamentos como de uma terapia gênica.
A síndrome de Pitt-Hopkins é caracterizada por déficit cognitivo, atraso motor profundo, ausência de fala funcional e anormalidades respiratórias, entre outros. Foi descrita em 1978, mas seu gene causador ficou conhecido apenas em 2007. Estima-se que a mutação no gene TCF4 ocorra em um a cada 35 mil nascimentos.
Minicérebros
Uma vez que a síndrome não se desenvolve em camundongos da mesma maneira que em humanos, o estudo em animais é inviável. Por isso, os pesquisadores usaram os chamados organoides cerebrais, um aglomerado de células humanas que cresce no laboratório e se assemelha a uma miniatura de cérebro em desenvolvimento, mas sem vascularização e com menos tipo celulares.
“O organoide cerebral é um modelo mais representativo do que qualquer outro para estudar disfunções do sistema nervoso central. Nesse caso, a célula obtida é do próprio paciente. Além disso, o organoide é tridimensional, portanto, seu funcionamento é mais próximo da realidade do que o de células cultivadas em placas, que crescem em apenas duas dimensões”, explica Papes.
Os organoides foram gerados a partir de biópsias da pele de portadores da síndrome, obtidas de pacientes recrutados na Unicamp e nos Estados Unidos, além de seus pais, que serviram como controle.
As células foram cultivadas para extrair os chamados fibroblastos, que são transformados em células-tronco pluripotentes, que, por sua vez, podem gerar muitos tipos de células humanas. Nesse caso, elas deram origem a neurônios, células progenitoras do sistema nervoso central e organoides cerebrais.
Enquanto as células dos pais dos pacientes formaram organoides que se desenvolviam normalmente, as dos portadores da síndrome cresciam menos, como resultado da menor replicação das células causada pela mutação e de um prejuízo da própria neurogênese. Ou seja, a geração de neurônios foi prejudicada por conta da mutação.
Além disso, os neurônios dos organoides com a mutação no TCF4 existiam em menor número e tinham menor atividade elétrica em comparação aos de organoides-controle. É sabido que a comunicação entre essas células é feita a partir de impulsos elétricos, sem os quais elas não podem exercer suas funções. Esse achado, portanto, pode explicar muitas características clínicas dos pacientes.
Os resultados foram semelhantes aos obtidos em tecidos do cérebro de um paciente com a condição, que faleceu por outras razões, o que reforça as conclusões obtidas com os organoides. O estudo foi o primeiro de que se tem notícia a estudar o cérebro de uma pessoa com síndrome de Pitt-Hopkins.
“O acesso ao cérebro post-mortem foi essencial para validarmos alguns dos resultados obtidos com os organoides cerebrais. O fato de termos visto características semelhantes entre o organoide criado em laboratório e o cérebro mostra o quão relevante é essa tecnologia”, afirma Muotri.
Terapia gênica
Uma vez observadas as alterações causadas pela mutação no gene TCF4, os pesquisadores buscaram maneiras de corrigi-la e, assim, realizar o que eles chamam de uma prova de conceito do que seria um tratamento.
Três intervenções foram testadas, uma delas utilizando a técnica de edição gênica conhecida como CRISPR-Cas9 – cujas criadoras ganharam o Prêmio Nobel de Química em 2020.
Para a estratégia envolvendo CRISPR, uma versão recente da técnica foi empregada para fazer com que a cópia funcional do gene existente na célula disfuncional passe a expressar muito mais proteína, compensando a cópia afetada pela mutação causadora da síndrome de Pitt-Hopkins.
Em outra intervenção, usando uma técnica diferente, os cientistas inseriram uma cópia extra do gene, que passou a exercer normalmente as funções gênicas, compensando a cópia mutada.
“Nosso genoma tem duas cópias de cada gene. O que causa a síndrome de Pitt-Hopkins é o fato de uma das cópias do TCF4 não funcionar. Inserir uma terceira cópia ou fazer com que a única cópia funcional expresse mais proteína para compensar a defeituosa pode solucionar o problema”, diz o pesquisador.
Os organoides que sofreram as intervenções passaram a crescer normalmente e tiveram um aumento da proliferação das células progenitoras, que no cérebro dão origem a diferentes tipos de célula, inclusive neurônios.
“Ainda que esse distúrbio seja considerado raro, existem outros que envolvem mutações nesse mesmo gene. Portanto, o que descobrimos aqui pode, futuramente, ser aplicado para transtornos como a esquizofrenia, por exemplo”, afirma Papes.
Uma terceira intervenção foi a aplicação de uma droga usada em estudos com células tumorais. Conhecida pela sigla CHIR99021, ela ativa uma via de sinalização celular conhecida como Wnt, muito estudada no contexto do câncer e que os autores descobriram ser alterada também por mutações no gene TCF4.
Em células e organoides disfuncionais tratados com a droga houve melhora em alguns indicadores moleculares e aumento de tamanho (no caso dos organoides). Os resultados abrem caminho para o desenvolvimento de medicamentos similares que possam tratar a disfunção, uma vez que a CHIR99021 ainda não pode ser utilizada em seres humanos.
“Essa via tratada com a droga é apenas uma das alteradas pela mutação no gene TCF4. A vantagem de uma terapia gênica em relação a um tratamento farmacológico é que ela resolveria o problema na sua origem. No entanto, a busca por novas drogas também é promissora”, diz Papes.
A pesquisa agora deve avançar para estudos pré-clínicos e clínicos. Os pesquisadores fecharam parceria com uma empresa especializada em terapia gênica, que está licenciando a tecnologia usada nos experimentos para que futuramente possa ser testada em humanos.
O trabalho teve ainda apoio da FAPESP por meio de bolsas de mestrado para José Ricardo Teixeira Júnior e de doutorado para Antônio Camargo, ambos pós-graduandos do IB-Unicamp.
A pesquisa teve também apoio do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), dos National Institutes of Health (NIH), nos Estados Unidos, e da Pitt-Hopkins Research Foundation.
O artigo Transcription Factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content pode ser lido em: www.nature.com/articles/s41467-022-29942-w.
